Vibronic spectra of molecules used to be hot research area of molecular science.
The intensity of the vibronic transition is governed by the Franck Condon principle
In this tutorial, we will use Gaussian 09 to calculate the vibronic transition intensity for a very small molecule (NH3). For larger molecules, the TD-DFT calculation of Hessian is very slow. But the user can try on their own supercomputer.
Second derivitive of the energy with respect to the molecular coordinates are very slow on my personal compute, with a Supercomputer, it will be feasible. And In Gaussian 09, the second derivative is done Numerically. There are 3N cartesian coordinates. For a centeral difference method (F’‘(x) = F(x+dx)+F(x-dx)-2F(x)/(2dx)) There are 6N calculations (plus one central coordinate x0). When the number of atoms in a molecular is getting large, it can be slow.
There are three input files for the Franck Condon Factor calculation in Gaussian
- The Geometry and Frequency / Normal Mode calculation of the Ground State
- The Geometry and Frequency / Normal Mode calculation of the Excited State (using TD-DFT or TD-HF or CIS etc)
- Calculation of Franck Condon Factor using results from 1 and 2.
As a result, in 1 and 2 check point file must be saved for step 3.
In the last step we will visualize the emission and absorption vibronic spetra of NH3
Ground State
Input
%NProcShared=4
%Chk=nh3_ground.chk
#P PW91PW91/6-311G(d,p) Opt(Z-Matrix) Freq PoP=Full
Title
0 1
N 0 0.93818 -0.02838 -0.07054
H 0 0.62658 0.80372 0.42833
H 0 0.62658 0.08845 -1.03367
H 0 1.95550 0.02404 -0.09618
Output
The Frequency section will look like this
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
A A A
Frequencies -- 1055.4278 1640.7715 1640.9691
Red. masses -- 1.1804 1.0648 1.0648
Frc consts -- 0.7747 1.6890 1.6894
IR Inten -- 149.9965 17.3644 17.3672
Atom AN X Y Z X Y Z X Y Z
1 7 0.00 0.00 0.12 0.03 -0.06 0.00 0.06 0.03 0.00
2 1 0.20 0.07 -0.53 -0.22 0.72 -0.04 0.17 -0.07 -0.26
3 1 -0.04 -0.21 -0.53 -0.49 -0.02 -0.20 -0.56 0.21 0.16
4 1 -0.16 0.14 -0.53 0.28 0.11 0.24 -0.43 -0.57 0.09
4 5 6
A A A
Frequencies -- 3384.1564 3508.9423 3509.1236
Red. masses -- 1.0269 1.0880 1.0880
Frc consts -- 6.9291 7.8929 7.8938
IR Inten -- 1.9437 0.4603 0.4606
Atom AN X Y Z X Y Z X Y Z
1 7 0.00 0.00 -0.04 0.07 -0.04 0.00 0.04 0.07 0.00
2 1 0.52 0.18 0.18 -0.50 -0.19 -0.22 -0.51 -0.15 -0.22
3 1 -0.11 -0.54 0.18 0.06 0.19 -0.08 -0.14 -0.71 0.30
4 1 -0.41 0.36 0.18 -0.54 0.49 0.30 0.16 -0.11 -0.08
First Excited State
Input
%NProcShared=4
%Chk=nh3_excited.chk
#P TD(Singlets) PW91PW91/6-311G(d,p) Opt(Z-Matrix) Freq=savenormalmodes PoP=Full
Title
0 1
N 0 0.93818 -0.02838 -0.07054
H 0 0.62658 0.80372 0.42833
H 0 0.62658 0.08845 -1.03367
H 0 1.95550 0.02404 -0.09618
Output
The frequency section will look like this
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
A A A
Frequencies -- 149.9133 957.7510 964.1658
Red. masses -- 1.2065 1.0368 1.0372
Frc consts -- 0.0160 0.5603 0.5681
IR Inten -- 24.7349 817.2925 804.3170
Atom AN X Y Z X Y Z X Y Z
1 7 0.00 0.00 0.12 -0.03 0.03 0.00 -0.04 -0.03 0.00
2 1 -0.01 0.01 -0.57 0.12 0.37 -0.01 0.69 0.18 0.00
3 1 0.01 0.00 -0.57 -0.30 -0.45 0.01 -0.13 0.60 0.01
4 1 -0.01 -0.01 -0.57 0.62 -0.41 0.00 -0.08 -0.33 -0.01
4 5 6
A A A
Frequencies -- 2128.5194 2133.5390 2447.5687
Red. masses -- 1.1669 1.1675 1.0079
Frc consts -- 3.1148 3.1311 3.5576
IR Inten -- 6063.8739 6049.2871 0.8808
Atom AN X Y Z X Y Z X Y Z
1 7 0.07 0.09 0.00 0.09 -0.07 0.00 0.00 0.00 0.00
2 1 -0.06 -0.43 -0.01 -0.37 0.57 0.02 -0.22 0.54 0.01
3 1 -0.41 -0.17 -0.01 -0.64 0.22 -0.02 0.58 -0.07 0.01
4 1 -0.47 -0.61 0.02 -0.20 0.17 0.00 -0.35 -0.45 0.01
Franck-Condon Factor Calculation
Input
%NProcShared=4
%Chk=nh3_ground.chk
#P Geom=AllCheck Freq=(ReadFC,FC,SaveNM) NoSymm
nh3_excited.chk
For Emission Spectra Calculation, just add EMI to the Freq Tuple.
Output
The start section of the result will look like this
**********************************************************************
Generation of the Franck-Condon spectrum
**********************************************************************
==================================================
Information on the Simulation
==================================================
Type of Spectroscopy: ONE-PHOTON ABSORPTION
Model applied to the transition: ADIABATIC HESSIAN
Approx. of the electronic transition dipole moment: FC
Temperature effect are not taken into account.
==================================================
Treatment of Input Data
==================================================
Data for initial state taken from current calculation.
Normal modes recovered from file.
Data for final state taken from checkpoint file "nh3_excited.chk"
Normal modes recovered from file.
Using excited electronic state number 1.
Initial state structure is set in Eckart orientation.
Final state structure is superposed to it.
Emission and Absorption Spectra of NH3
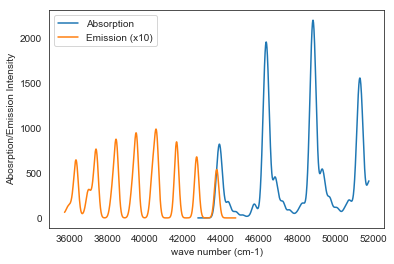
Since the emission intensity is too small, we multiplied it by a factor of 10. It can clearly be seen that there is a wave length shift between the emission and absorption spectra. This shift is called Stokes Shift which is due to the shift in equilibrium geometry between the ground and excited states.